造礁珊瑚受到海洋变暖的严重威胁,需要积极干预以减少珊瑚白化和死亡率。珊瑚与各种各样的细菌有关,这些细菌会影响珊瑚的健康,但对珊瑚在热压力下可能有益的具体功能的了解却很少。根据珊瑚白化的氧化应激理论,清除活性氧(ROS)或氮种(RNS)的细菌有望增强珊瑚的热恢复能力。此外,细菌的碳输出可能取代藻类光共生体的碳供应,增强热恢复能力,促进漂白恢复。为了鉴定候选益生菌,我们对82种从新兴珊瑚模型星系束状菌中分离出来的纯培养细菌的基因组进行了测序。
基因组分析显示分离的细菌隶属于37个属。分离菌株如Ruegeria、Muricauda和Roseovarius被发现编码合成抗氧化剂甘露醇、谷胱甘肽、二甲基硫化物、二甲基磺酰丙酸、玉米黄质和/或β-胡萝卜素的基因。在许多束状菌相关细菌中发现了参与rna清除的基因,这是几个属(包括假噬杆菌)的新发现。在包括假弧菌和Roseibium在内的7株菌株中检测到可能输出碳的转运蛋白(semi - weet)。此外,一系列细菌菌株,包括玫瑰属和玫瑰属菌株,揭示了可能增强细菌与珊瑚宿主定植和关联的基因组特征,如分泌系统和真核样重复蛋白。
我们的工作提供了对G. fascularis相关细菌功能潜力的深入基因组分析,并确定了可能增强珊瑚抵御珊瑚白化能力的新性状组合。识别和表征对珊瑚有益的细菌对于开发有效的益生菌至关重要,益生菌可以提高珊瑚的气候适应能力。
视频摘要
造礁珊瑚和珊瑚礁正受到气候变化迫在眉睫的威胁。在气候变化驱动的夏季热浪期间,海面温度升高加上高辐射水平经常发生,这是大规模珊瑚白化的主要原因[1]。珊瑚白化是指珊瑚宿主与共生科藻类共生体之间的专性共生关系被破坏,导致共生体与珊瑚宿主组织分离。这使得寄主处于碳剥夺状态[2],随之而来的往往是珊瑚死亡和珊瑚礁退化。越来越多的人担心,海洋变暖的速度太快,珊瑚的自然适应能力跟不上,因为它们的繁殖周期相对较长。这一概念导致了一个新的研究领域,旨在加速进化过程以增强珊瑚的白化恢复能力[3]。珊瑚辅助进化的概念包括,除其他方法外,操纵与珊瑚有关的微生物共生体,如细菌。珊瑚相关细菌是珊瑚健康和适应性的重要参与者,因为它们通过合成抗菌化合物[4]保护珊瑚宿主免受病原体的侵害,产生抗氧化剂和循环营养物质,如氮、硫、碳和磷[5]。珊瑚相关细菌群落组成与珊瑚耐热性之间的相关性表明,细菌在珊瑚耐热性中起着有益的作用[6],但潜在的机制目前尚不清楚。微生物组操作已成功应用于农业和医学等领域[7],但在刺胞动物中仍处于起步阶段。尽管如此,珊瑚微生物组操纵的可行性最近已经得到证实[8,9]。此外,最近在治疗珊瑚白痘病[10]和石珊瑚组织损失病[11]方面取得了成功。旨在通过微生物组操作提高珊瑚白化恢复能力的研究也显示了积极的结果,尽管添加的细菌是否以及如何推动对热应激的耐受性的提高仍有待探索。这些研究用从珊瑚和海水中分离的细菌接种珊瑚,并测试了潜在的有益功能(对病原体的抗菌活性、抗氧化酶过氧化氢酶的活性以及负责硫和氮循环的基因的存在),并证明了damicornis Pocillopora[12]减少了热漂白和对病原体感染的表型反应,并增强了hispida Mussismilia的漂白恢复[13]。另一项研究表明,用耐热同种虫的整个微生物群接种热敏poillopora sp.和Porites sp.后,它们的漂白耐受性增强[14]。
漂白的主要理论是氧化应激假说,该假说认为温度和光照的升高会损害光共生体的光系统II、还原性五糖磷酸循环反应和类囊体膜[15,16],从而导致有毒活性氧(ROS)的过量产生[17]。这可能会压倒抗氧化反应,过量的ROS扩散到珊瑚宿主细胞中,在那里它们会对大分子(例如DNA)造成损害,并引发细胞级联,导致漂白[17,18]。即使在非胁迫条件下,symbiodiiaceae植物在光合作用过程中也会不断产生各种ROS,如单线态氧(1O2)、超氧化物(O2?)、过氧化氢(H2O2)和羟基自由基(OH?)[19],并被光共生体和宿主细胞中由各种酶和非酶机制组成的抗氧化防御系统迅速清除。清除酶包括过氧化氢酶和超氧化物歧化酶,非酶抗氧化剂包括甘露醇、谷胱甘肽和类胡萝卜素[20]。除活性氧外,活性氮(reactive nitrogen species, RNS),如一氧化氮,也可能参与珊瑚白化[18,21]。共生菌科(symbiodiiiaceae)培养物和海葵(Exaiptasia diaphana)中一氧化氮水平的升高,以及共生菌科(symbiodiiaceae)中一氧化氮生成酶活性的升高与刺胞菌白化和热胁迫有关[22,23,24,25]。一些研究表明,在白化过程中,一氧化氮在诱导宿主凋亡途径中发挥作用,以应对共生功能障碍[25,26,27,28]。ROS和RNS的信号通路也可能相互作用[18,22,26]。其中一种相互作用是O2?和一氧化氮产生过氧亚硝酸盐(ONOO?),这会破坏线粒体内的电子传递[21],并与共生二科植物的热应激有关[26]。
氧化应激理论认为,由光和温度胁迫引起的ROS和RNS的过量产生触发了细胞级联反应,导致白化。除了氧化应激理论外,一些研究假设,白化级联反应是由于宿主在温度升高的情况下无法为生长更快的共生菌科提供足够的二氧化碳而引发的[29,30]。这被认为会破坏Calvin-Benson循环,导致共生菌过量产生活性氧,而活性氧可能会泄漏到宿主细胞中,引发漂白级联反应。第三种理论认为,温度升高会影响珊瑚与其共生藻类之间的营养循环[2]。热应激增加宿主呼吸和分解代谢过程,导致铵成为共生菌科可利用。因此,共生体科从通常的氮限制状态中解放出来,允许它们的生长增加,并导致它们利用大部分光合产物为自己的生长而不是将其转移到珊瑚宿主。共生体科植物很快就会耗尽磷,随之而来的N:P失衡被认为会导致其类囊体膜组成的变化,损害光系统[31],再次产生过量的活性氧,这可能会导致共生体科植物从珊瑚宿主中流失。
基于活性氧和RNS在珊瑚白化中的作用,中和这些分子的机制可能会提高珊瑚的耐热性。事实上,当通过添加外源性抗氧化化合物降低ROS水平时,珊瑚和珊瑚模型动物(海葵)的白化程度会降低[32,33],并且一种一氧化氮清除化合物介导了热胁迫下symbiodiiaceae光合效率的降低[25]。我们假设,通过微生物组操作来增强珊瑚全息生物体内ROS和rns的降解,例如给珊瑚接种具有高ROS和/或rns清除能力的细菌,可能是一种有用的保护策略。虽然这可能会减少藻类共生体的排出,但由于共生体科的呼吸速率较高和碳转运较低,珊瑚宿主可能仍然是碳限制的。因此,具有将碳转运到宿主的额外潜力的ROS和rns清除细菌可能通过减少宿主饥饿提供额外的好处[34]。因此,糖转运体的存在可以将碳从细菌细胞中转运出来,这可能是一个有益的特性。在植物和其他真核生物中发现的“糖最终会被出口的转运体”(SWEET)[35,36]可以双向运输小糖分子,特别是葡萄糖[37]。半甜蛋白是甜蛋白的细菌同源物。
在这里,我们确定了细菌益生菌候选物来减轻硬核珊瑚束状星系的热应激,这是一种新兴的珊瑚模型[38]。我们分析了82株分离自束状螺旋体的纯培养菌株的基因组序列,重点研究了与ROS和rns清除有关的性状以及糖输出机制。此外,我们调查了一系列其他可能表明稳定宿主关联的潜在有益代谢途径和基因组特征,例如在互惠内共生生物中发现的分泌系统,已知可促进逃避真核宿主免疫系统。此外,真核样重复蛋白(ELPs),包括微生物锚蛋白重复序列(ARPs)和wd40重复序列,被认为可以促进宿主感染,并通过协助蛋白-蛋白相互作用促进稳定的共生[39,40]。
从澳大利亚大堡礁的萨德伯里礁(S -17°01 E 14°21)收集了三群珊瑚G. fascicularis,将其带入凯恩斯海洋(昆士兰凯恩斯)的水族馆设施,然后于2020年2月运往墨尔本大学。抵达后,珊瑚在140升的循环水缸系统中过夜,该系统含有26±1.1°C,盐度为35±0.5 ppm (ppt)的反渗透水重组红海盐?(RSS, R11065, Red Sea, USA)。为了进行细菌培养和16S rRNA基因元条形码编码,使用水牙线(Waterpik,澳大利亚)将每个菌落随机选择的30个息肉的珊瑚组织和粘液取样于100 mL过滤消毒(0.2μm) RSS水(fRSS)中。组织匀浆在无菌Falcon管中3750 rcf离心10分钟,所得组织微球转移到1.5 ml无菌Eppendorf管中,在5000 rcf离心10分钟。去除上清后,组织微球在1 ml fRSS中匀浆,使用30 Hz的组织裂解器(Tissue- lyser II, Qiagen, Chadstone, Australia)。然后,对每个菌落的每个匀浆进行连续稀释,从10?2到10?7。剩余未稀释的组织匀浆液(4倍200μL)转移至1.5 ml无菌Eppendorf管中,在液氮中快速冷冻,保存于- 80°C,待进一步处理16S rRNA基因元条形码。将每种稀释液各50微升平板接种于三个培养皿中(R2A琼脂CM0906, Oxoid Ltd.)。Basingstoke, Hampshire, England),补充40 g L?1 fRSS,并在MA (Difco?Marine Agar 2216, BD, Sparks, MD, USA)的三个培养皿上。培养皿在黑暗中孵育,温度为26℃,与采集地点的季节性海水温度相似。培养1周后,采摘单个菌落,每个菌落在新培养基上继代培养至纯度。单个菌落(可能由一株细菌组成)在200μL 40%甘油中重悬,并在- 80°C保存。
为了鉴定g . fascularis来源的分离株,新鲜生长的细菌菌落在20μL milliq水中溶解,95°C, 2000 g离心1分钟,上清液作为DNA模板进行后续菌落PCR扩增。使用细菌引物27f (50- AGA GTT TGA TCM TGG CTC AG-30)和1492r (50- TAC GGY TAC CTT GTT ACG ACT T-30)进行菌落pcr[41],遵循先前描述的方案[42],但修改热循环如下:在94°C下5分钟;30个循环,94℃下60 s, 50℃下45 s, 72℃下90 s;72℃下10min;最终保温温度为4°C。通过1%琼脂糖凝胶电泳验证16S rRNA基因扩增子的产生后,对PCR产物进行纯化,并在Macrogen (Seoul, South Korea)使用引物1492r在ABI测序机上进行Sanger测序。在geneprime 2021.1.1 (https://geneious.com)中对原始序列进行对齐和修剪,并通过BLASTN (https://blast.ncbi.nlm.nih.gov)将校正后的16S rRNA基因序列(~ 1000 bp)与GenBank序列进行比较,以找到最高百分比的同源性。
为了评估细菌分离物在多大程度上代表了完整的束状螺旋体细菌微生物组,我们对16S rRNA基因进行了扩增子测序。我们根据海洋微生物共生实验室(the University of Melbourne)的既定方案[43],从每个G. fascularis珊瑚群落的三个组织匀浆样本中提取DNA,并纳入不含样本的阴性对照(n=3),以确定DNA提取中的污染物。提取的DNA和阴性对照(3个DNA提取对照,3个PCR对照)一式扩增,并建立测序文库,两者都遵循既定的协议[44]。文库在墨尔本大学Walter and Eliza Hall医学研究所(WEHI, University of Melbourne)使用v3 (2 × 300 bp)试剂进行Illumina Miseq测序。
测序结果显示,每个样本的平均读数为40,786。在QIIME2 v2021.4中对16S rRNA基因序列进行了处理和解复用[45]。使用Cutadapt v2.6去除引物[46]。DADA2[47]在QIIME2环境中进行质量控制和筛选步骤,包括测序错误的纠正、低质量碱基(平均Qscore < 30)的去除和扩增子序列变异(amplicon sequence variants, asv)的产生。质量控制和过滤步骤导致每个样本有20,244个reads,总共确定了3154个asv。使用SILVA v138数据库对每个ASV进行分类[48]。在R[49]中使用phyloseq[50]和microbiome[51]包分析了16S rRNA基因元条形码数据集的多样性指标。
从91个细菌分离株中选择82个进行全基因组测序,目的是尽可能多地覆盖分类多样性。根据制造商的方案,使用Invitrogen?Purelink?基因组DNA迷你试剂盒(K182001, Invitrogen?,Thermo Fisher Scientific,澳大利亚)提取新鲜培养的细菌基因组DNA,但包括以下修改:在溶菌酶消化缓冲液中加入10 mg mL?1溶菌酶,将细胞颗粒在溶菌酶消化缓冲液中重悬后,将样品超声处理2 min (POWERSonIC 505 Digital Ultrasonic Bath, Thermoline Scientific, Australia),在55°C下孵育后,加入20μL RNase(试剂盒提供)。使用Quant-iT?PicoGreen?dsDNA检测试剂盒(P7589, Invitrogen?,Thermo Fisher Scientific,澳大利亚)评估DNA浓度,遵循制造商的指导方针,在485 nm激发和530 nm波长下测量其吸光度(CLARIOstar PLUS板读仪,BMG Labtech,澳大利亚)。用UV5Nano (Mettler Toledo, Australia)测定提取DNA的质量。在准备全基因组测序时,将每个细菌分离物提取的基因组DNA浓度调整为20 ng/μL。多重Nextera XT (Illumina)文库在Doherty应用微生物基因组学(Doherty Institute, University of Melbourne, Australia)的Illumina NextSeq550平台上测序,产生150 bp的配对末端reads。
使用Trimmomatic v0.39对原始reads进行修剪和质量过滤(去除前10个碱基,reads < 30 bp和Phred评分≤28的reads,平均为4 bp)[52]。修剪前后的原始读数质量用FASTQC v0.11.5[53]检查,用MultiQC v1.12[54]整理。使用SPAdes基因组组装器3.11.1[55]重新组装裁剪过的和经过质量过滤的reads,分别含有21、33、55和77 k-mers,并使用“-谨慎”选项将错配和短索引的数量降至最低。使用BBMap v38.96[56]删除所有长度< 1000 bp的contigs。随后,使用CheckM v1.16评估组装基因组的完整性、异质性和污染水平[57]。基因预测在Prokka v1.14.6[58]中进行,使用prodigal v2.6.3[59],参数为“-addgenes”,“-addmrna”,“-rfam”。预测的蛋白质使用Interproscan 5.55 v88.0[60]搜索,使用参数“- apply Pfam”和值< 1e?5注释蛋白家族(Pfam) id。在基因组分类数据库(Genome Taxonomy Database, GTDB- tk v2.1.0)[61]上使用“classify_wf”工作流对所有组装的基因组进行分类分配[62]。GTDB-Tk基于120个细菌标记基因对基因组进行分类。在IQ树v1.6.1[63]中,采用最大似然方法,采用ModelFinder Plus在IQ树中实现的LG + F + R7模型(一般矩阵(LG),经验基频(F), 7类FreeRate模型(R7), AIC 424879.963, BIC 46067.398))作为最佳模型,并利用GTDB-Tk生成的120个细菌标记基因的多个序列比对的1000个超快bootstrap,构建了系统发育树。该树在iTOL v6中可视化[64]。
使用FastANI v1.33[65]、FastAAI (https://github.com/cruizperez/FastAAI)和GenDisCal v1.3.1 (https://github.com/LM-UGent/GenDisCal)的默认参数分别计算基因组特征,包括平均核苷酸同一性(ANI)、平均氨基酸同一性(AAI)和计算机基因组-基因组距离(GGD)。所有描绘ANI, AAI和GGD的矩阵都在R v1.4.17[49]中绘制,使用软件包“ggplot2”v3.3.5[66]。代谢途径、运输系统和分泌系统通过metabolic - g v4.0[67]注释,应用“-m-cutoff 0.50”来包括≥50%完成的途径。代谢途径(KEGG模块数据库,https://www.genome.jp/kegg/module.html)、转运体和分泌系统的完整性在enrichment m v0.6.4中进行了估计[68]。使用R中的ggplot2 v3.3.5[66]软件包进行绘制(补充图2)。使用Interproscan 5.55 v88.0[60]使用Pfam id对不同类别的elp和其他感兴趣的基因进行注释(表1)。所有Pfam识别代码均来自Uniprot数据库(http://k1.fpubli.cc/file/upload/202308/30/vniq4f4esoo)。ARP和WD40 ELP家族的存在是根据它们的Pfam识别码确定的(表1)。SemiSWEET转运蛋白也通过它们的Pfam识别码检测到(表1)。我们使用MUSCLE v3.8.425[69]在genous Prime v2021.1.1中比对了分离物中发现的每个SemiSWEET转运蛋白的蛋白质序列。采用ModelFinder Plus选择的mtZOA + G4模型(线粒体后处理动物蛋白模型(mtZOA),伽马率异质性(G), AIC 3359.763, BIC 3407.600),在iTOL v6中可视化,在IQ树v1.6.1[63]中构建了半isweet蛋白序列的系统发育树。根据pam识别码,还鉴定了多个参与ROS和rns清除的基因(表1)。一个参与ROS清除的基因包括gshAB(双功能谷氨酸-半胱氨酸连接酶gshA/谷胱甘肽合成酶gshB),该基因负责抗氧化剂谷胱甘肽的生物合成。通过番茄红素β-环化酶(crtY)基因检测抗氧化剂β-胡萝卜素的合成,通过β-胡萝卜素羟化酶(crtZ)基因检测抗氧化剂玉米黄质的合成。通过甘露醇-1-磷酸脱氢酶(mtlD)和甘露醇- 2-脱氢酶(mtlk)测定编码甘露醇生物合成的基因。通过超氧化物歧化酶(SOD)的存在,确定了酶的超氧化物清除潜力。通过二甲基磺酰丙酸(DMSP)裂解酶(dddQ, dddY, dddL)的存在确定了抗氧化剂二甲基硫化物(DMS)的生物合成,通过dsyB(甲基硫羟基丁酸甲基转移酶)的存在确定了抗氧化剂DMSP的生物合成。清除不同形式RNS的潜力,如过氧亚硝酸盐的还原,是从过氧化物还原素(ahpC)的存在推断出来的。通过两个不同的基因,hmp(一氧化氮双加氧酶)和norBC(一氧化氮还原酶亚基C (norC)和B (norB))来评估一氧化氮的清除能力。我们还筛选了负责将一氧化二氮(norBC的产物)转化为氮并完成反硝化的基因nosZ(一氧化二氮还原酶)。
表1基因和各自的蛋白家族(Pfam id)g . fascicularis-相关细菌基因组被询问。选择感兴趣的Pfam id基于已知的它们通过酶活性或关键抗氧化剂的产生参与ROS和rns清除,通过双向糖转运通过真核重复蛋白的Nal糖转运体和珊瑚-细菌共生的建立和促进。感兴趣的Pfam id来自Uniprot数据库se (http://k1.fpubli.cc/file/upload/202308/30/vniq4f4esoo。为了标注感兴趣的Pfam id,使用Interproscan 5.55 v88.0 [60]
摘要
介绍
材料与方法
结果
讨论
结论
数据和材料的可用性
参考文献
致谢
作者信息
道德声明
补充信息
搜索
导航
#####
我们总共从三个大堡礁(GBR)菌落的组织和粘液中培养了613株分离细菌,跨越54属91种(补充表1)。每个分离细菌首先通过16S rRNA基因的Sanger测序和从Basic Local Alignment Search Tool (BLASTn)中选择最接近的匹配进行分类鉴定[70]。大多数菌株属于γ变形菌纲(235株,补充表1),其次是α变形菌纲(124株)、黄杆菌纲(27株)和芽孢杆菌属(180株)、Alteromonas(153株)、Vibrio(70株)和Ruegeria(50株)。这些细菌分离物所属属占三个G. fascularis菌落细菌群落相对丰度的48%(图1,Supplementary Fig. 1)。重要的是,17个最丰富的扩增子序列变异(asv)所属属中有70%是在纯培养中获得的,包括Ruegeria和Alteromonas。通过元条形码鉴定出的细菌属是用于培养的G. fascularis菌落中最丰富的asv,但未在培养中获得,包括Endozoicomonas和Thalassotalea。选取相对丰度> 2%的17个最丰富的asv。
图1
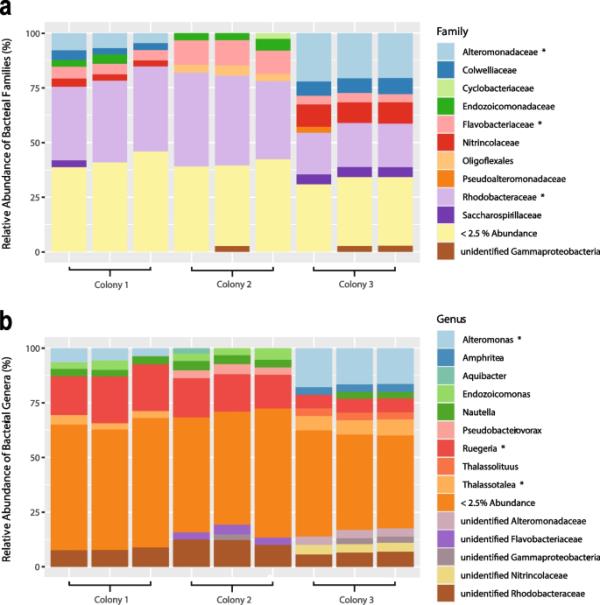
菌落的细菌群落组成(基于16S rRNA基因元条形码)突出了每个菌落中每个重复样本中最丰富的细菌科和最丰富的细菌属(n=3个/珊瑚菌落;SILVA数据库v. 138)。三个最丰富的科/属用星号突出显示。较少的细菌科或属被归为一类(相对丰度< 2.5%)。
我们对包括红杆菌科(GTDB-Tk分类,26株)和Alteromonadaceae(11株)在内的16个细菌科的82株菌株进行了基因组测序和汇编(图2)。总体而言,通过GTDB-Tk使用120个细菌标记基因鉴定了37个不同的细菌属(补充表2)。收集中最丰富的属是Alteromonas(10株),Ruegeria(10株),芽孢杆菌(8株),弧菌(8株)和齐蓬源菌(5株,basionym)。通过GTDB-Tk与NCBI中最接近命中的菌株的16S rRNA基因序列的分类定位比较,细菌基因组的分类归属较少(82株菌株中有37属)(120个标记基因;补充表2)与91个分离株的54属(16SrRNA基因;补充表1)。这种差异源于GTDB和16S rRNA基因序列(最接近NCBI中的BLASTn)在某些情况下分配的属不同,以及GTDB和NCBI在分类学命名上的微小差异。
图2

82株纯种培养的束状葡萄球菌相关细菌的系统发育树。82株分离菌分属16个细菌科。除了5个基因组外,所有基因组都组装了16S rRNA基因(粉色星星)。利用基因组分类数据库(Genome Taxonomy Database, GTDB- tk v2.1.0)[61],通过GTDB- tk v2.1.0[62]对120个细菌标记基因进行细菌基因组的分类。采用IQ tree v1.6.1[63]中的最大似然法构建系统发育树,ModelFinder Plus选择LG + F + R7模型为最佳模型,利用120个细菌标记基因的多个序列比对的1000次超快bootstrap构建系统发育树。该树在iTOL v6中可视化[64]。多尔蒂微生物基因组学
所有82个组装的细菌基因组的平均完整性为99.59±0.45%,污染为0.46±0.41%(补充表2,补充图4-6)。这些基因组的平均大小为4.34±0.86 Mb,平均组装成54.47±41.57个contigs,编码4071.96±754.07个基因,编码密度为89.57±1.96%。所有基因组的鸟嘌呤-胞嘧啶(GC)含量平均为51.67±9.88%,在77个基因组中组装了16S rRNA基因(图2)。
我们对带注释的基因组进行了筛选,以寻找在ROS和rns清除中起作用的基因(表1、图3、补充表2)。在不同的细菌科中发现了与谷胱甘肽、甘露醇和DMSP合成相关的基因,而玉米黄质、β-胡萝卜素和DMS合成相关的基因,和超氧化物清除仅限于两到三个细菌家族(图3)。在大量不同的家族中检测到所有具有rna清除功能的感兴趣基因(图3)。
图3
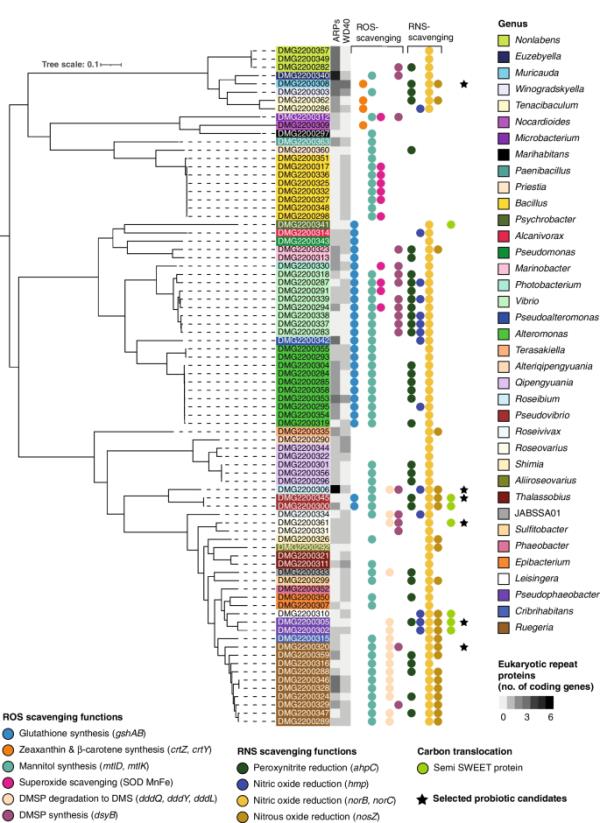
在82株G. fascularia相关细菌的系统发育树中发现了ROS和rns清除基因、semi - weet蛋白和真核重复序列蛋白(ARPs和WD40-repeats)。分离得到的82株细菌隶属于37个细菌属。选定的候选益生菌用黑色星号表示。利用基因组分类数据库(Genome Taxonomy Database, GTDB- tk v2.1.0)[61],通过GTDB- tk v2.1.0[62]对120个细菌标记基因进行细菌基因组的分类。采用IQ tree v1.6.1[63]中的最大似然法构建系统发育树,ModelFinder Plus选择LG + F + R7模型为最佳模型,利用120个细菌标记基因的多个序列比对的1000次超快bootstrap构建系统发育树。该树在iTOL v6中可视化[64]。多尔蒂微生物基因组学
在一种或多种细菌分离株中发现完成度≥80%的代谢途径(KEGG途径模块)包括氨基酸代谢、萜类和多酮类的生物合成、碳水化合物代谢、能量代谢、聚糖代谢、脂质代谢、辅助因子和维生素的代谢以及核苷酸代谢(补充图2、补充表3,补充结果文本“在G. fascularis -associated bacteria中发现的完整代谢途径”)。与能量代谢(如碳代谢)相关的途径在几个分离株中被发现是100%完整的。例如,还原五糖磷酸循环(KEGG模块ID M00167)在Rhodobacteraceae(25株),Sphingomonadaceae(5株)和Stappiaceae (3, Supplementary Fig. 2, Supplementary Table 3)中被证明是100%完整的。另一个参与碳代谢的途径,磷酸乙酰转移酶-醋酸激酶途径(M00579)在Rhodobacteraceae(13株),Alteromonadaceae(9株),Bacillaceae(9株),Vibrionaceae (9), Microbacteriaceae (1), Moraxellaceae (1),油茶科(1)、Paenibacillaceae(1)和假单胞菌科(1),只有4株菌株含有无氧光系统II (M00597),即Roseibium sp.菌株Doherty Microbial Genomics (DMG)2200306, Roseivivax marinus (DMG2200334)和2株Roseovarius sp. (DMG2200331, DMG2200361)。参与氮代谢的途径,如反硝化(M00529),在红杆菌科(10株),葡萄球菌科(3株)和Terasakiellaceae(1株)中100%完整。SemiSWEET蛋白的基因存在于7株细菌中(图3):Psychrobacter sp. (DMG2200341), 2株反硝化假弧菌(DMG2200345, DMG2200300), Roseovarius sp. (DMG2200361), Leisingera sp. (DMG2200310)和2株假噬杆菌sp. (DMG2200305, DMG2200302)。基于semiweet蛋白比对的系统发育分析反映了鉴定出它们的细菌分离株的系统发育(图4)。
图4
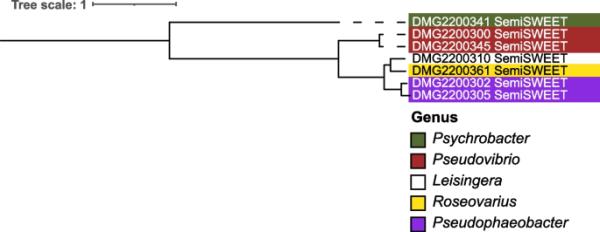
束状菌相关细菌中发现的SemiSWEET蛋白序列系统发育树。利用MUSCLE v3.8.425[69]在genisprime v2021.1.1中对各半weet转运蛋白序列进行比对。采用IQ tree v1.6.1[63]中的最大似然法构建系统发育树,ModelFinder Plus选择mtZOA + G4模型为最佳模型。该树在iTOL v6中可视化[64]。多尔蒂微生物基因组学
在ELP蛋白中,我们发现ARPs比WD40-repeat蛋白更丰富(图3)。总的ARPs数量最多的是Roseibium sp. (DMG2200306, 6个ARPs)和Euzebyella sp. (DMG2200340, 5个ARPs)。在Muricauda sp.中检测到最多的WD40-repeat蛋白(DMG2200308, 3个WD40-repeat)。100%完整的分泌系统包括II型(T2SS), III型(T3SS), IV型(T4SS)和VI型(T6SS,补充图3)。T2SS在5个科中有很好的代表,即Alteromonadaceae(11株),Vibrionaceae(9株),Oleiphilaceae(2株),Pseudomonadaceae(1株)和Alcanivoraceae(1株)。T3SS仅在弧菌科的5株中检测到,在Vibrionaceae中检测到T4SS,在Vibrio harveyi弧菌中检测到T4SS,在Rhodobacteraceae中检测到T4SS,在Vibrionaceae (7), Stappiaceae (3), Oleiphilaceae(2)。Alteromonadaceae(1)和Pseudomonadaceae(1)。
我们通过优先考虑编码ROS和/或清除rns的基因的存在,然后是semiSWEET转运体的存在,选择了六种候选益生菌(图3,黑色星星)。其他选择标准包括基因组特征,即elp (ARPs和wd40重复序列)和分泌系统,这些特征被认为可以增强细菌与珊瑚宿主的定植和联系。我们从益生菌选择中排除了弧菌科菌株,因为已知该家族含有几种珊瑚病原体[71]。
1个候选者,Ruegeria sp. (DMG2200320;GTDB-Tk分类;在NCBI中,98.03%的16S rRNA基因序列与R. marisrubri菌株ZGT118一致,显示了与甘露醇(mtlD, mtlK), DMS (dddQ)和DMSP合成(dsyB,图3)有关的基因,包括甘露醇转运体(M00200,补充表3)。编码一氧化氮还原的两个感兴趣的基因(hmp, norBC)也在Ruegeria sp. DMG2200320中被发现,有助于完整的反硝化途径(M00529,补充图2)。DMG2200320没有显示任何分泌系统,但有一个ARP(图3)。完整的钴胺素(维生素B12)合成途径也存在(M00122,补充图2,补充表3)。
第二个候选益生菌是Muricauda sp. (DMG2200308;GTDB-Tk分类;99.04%的16S rRNA基因序列与NCBI中的ruestringensis DSM 13258一致,其中包含玉米黄质和β-胡萝卜素合成(crtZ, crtY)以及过氧亚硝酸盐(ahpC),一氧化氮(norBC)和氧化亚氮(nosZ,图3)的基因。DMG2200308还显示了三个ARPs和WD40-repeat蛋白,这是所有分离株中记录到的WD40-repeat蛋白的最高数量(图3)。
第三个候选益生菌是Roseibium sp. (DMG3300306;GTDB-Tk分类;NCBI中与R. album菌株5OM6 16S rRNA基因序列同源性为98.91%;同型同义词白Stappia[72]和Labrenzia alba[73]),包含甘露醇(mtlD, mtlK), DMS (dddQ)和DMSP (dsyB)合成基因(图3)和甘露醇转运体(M00200,补充表3)。我们还检测了一氧化氮(norBC, hmp)和氧化亚氮(nosZ,图3)的还原基因,以及完整的反硝化途径(M00529,补充图2,补充表3)。在所有分离物中,DMG3300306具有最多的ARPs(6个ARPs)。一个WD40-repeat蛋白(图3)和一个完整的T6SS(补充图3)。
第四个候选益生菌是Roseovarius sp. (DMG2200361;GTDB-Tk分类;在NCBI中,与aestuarii菌株SMK-122的16S rRNA基因序列一致性为98.8%,包含DMSP合成基因(dsyB)和一氧化氮还原基因(norBC,图3)。DMG2200361还含有一个半糖糖转运蛋白(图3)。在Roseovarius sp. DMG2200361中检测到一个ARP和WD40-repeat蛋白(图3),以及一个完整的T4SS(补充图3)。
第五种选定的候选益生菌是假噬杆菌sp. (DMG2200305;GTDB-Tk分类;与莱辛era methylohalidivorans DSM 14336菌株MB2在NCBI中具有96.1%的16S rRNA基因序列一致性,是唯一显示所有清除rns基因(ahpC, hmp, norBC, nosZ)和合成DMS的dddQ的菌株(图3)。我们还在菌株DMG2200305中发现了一个半weet转运体(图3)。我们没有在假噬杆菌sp. DMG2200305中发现任何elp,但发现了一个完整的T4SS(补充图3)。
反硝化假弧菌(DMG2200345;GTDB-Tk分类;与NCBI中P.反硝化菌菌株DN34的16S rRNA基因序列100%一致,是我们选择的第6个候选益生菌,具有合成抗氧化剂谷胱甘肽(gshAB)和甘甘醇(mtlD, mtlK,图3)的基因。DMG2200345中清除rns的基因包括过氧亚硝酸盐(ahpC)、一氧化氮(nosBC)和氧化亚氮(norZ)还原(图3),以及完整的反硝化途径(M00529,补充图2)。补充表3)。我们在P.反硝化菌DMG2200345中发现了一个半weet转运蛋白(图3)和一个T6SS,但没有发现任何elp(补充图3)。
为了鉴定可能有助于建立珊瑚白化恢复能力的候选益生菌,我们通过基因组筛选研究了星系束相关细菌的功能潜力。我们发现了与束状丝状菌相关的细菌具有各种新型的有益功能组合,如ROS和/或rns清除,可以减轻珊瑚全息体中的热应激,碳转运可能有助于抵抗漂白和恢复,以及基因组特征表明可以增强细菌定植和与珊瑚全息体的关联。在G. fascularis中,ROS被认为是热漂白的主要驱动因素[74],所选择的益生菌候选菌可以中和ROS,减少或防止这种珊瑚物种的漂白。我们选择的细菌益生菌候选者也可能与广泛的其他珊瑚物种相关,因为它们是在硬核珊瑚中常见的分类群。此外,已知G. fascicularis在世界范围内广泛分布于各种珊瑚礁环境中,甚至是一些近海边缘珊瑚礁的优势物种[75]。
获得的82株细菌基因组包括16科37属。这些家族构成了与三个gbr来源的束状菌菌落相关的细菌微生物组的大部分。16S rRNA基因元条形码鉴定出的细菌属中,鲁氏菌属(平均相对丰度为11.68%)和交替单胞菌属(平均相对丰度为7.73%)丰度最高。在G. fascularis细菌微生物组中发现的细菌,但我们无法培养的细菌包括内植单胞菌(Endozoicomonas)等属,内植单胞菌作为珊瑚健康的潜在指标受到越来越多的关注[76],并且已被证明通过将DMSP代谢为DMS在珊瑚硫循环中发挥作用[77]。总体而言,GBR菌落的微生物组与来自南海的G. fascicularis相似[78,79]。当我们将研究重点扩大到一般硬核珊瑚的细菌微生物组时,我们发现培养收集的所有细菌类别(即Gammaproteobacteria, Alphaproteobacteria, Bacilli和Flavobacteriia)都与珊瑚有关[80,81]。总的来说,我们收集的细菌基因组是与G. fascicularis和硬核珊瑚相关的细菌多样性的全面代表。
所选的三种候选益生菌,即Ruegeria sp. DMG2200320, Roseibium sp. DMG3300306和Roseovarius sp. DMG2200361,显示出产生两种抗氧化剂DMS和DMSP的潜力[82];伪噬杆菌sp. DMG2200305含有仅用于生产DMS的基因。通过DMSP的去甲基化或裂解合成DMS的方法在假噬杆菌(Pseudophaeobacter sp.)[83]、鲁氏杆菌(Ruegeria sp.)[84,85]和玫瑰病菌(Roseovarius sp.)[86]中已有报道,而通过dsyB合成DMSP对后两属来说是全新的。DMS和DMSP的合成都是已知的Roseibium sp.[87,88]。一般来说,DMSP和DMS都被认为是清除OH -的有效抗氧化剂,DMS是活性更强的化合物[82]。DMSP和DMS还可以作为微生物的碳和硫源[89],或通过抗菌特性塑造珊瑚微生物群落[90]。DMSP还可以作为珊瑚病原体珊瑚弧菌(Vibrio coralliilyticus)在珊瑚宿主上定植的化学引诱剂[91]。我们在三种候选益生菌(Ruegeria sp. DMG2200320, Roseibium sp. DMG3300306, Pseudovibrio反硝化菌DMG2200345)中检测到合成抗氧化剂甘露醇的潜力,这是对Ruegeria sp.和P.反硝化菌的新观察。甘露醇清除OH?[92],可以减轻珊瑚的热应激。例如,在珊瑚中外源添加甘露醇可以减少热胁迫下Agaricia tenuifolia中Symbiodiniaceae的损失[32],也可以减少热胁迫下Pavona divaricate宿主组织中的DNA损伤[93]。它还减轻了E. diaphana的漂白[94]。在候选益生菌Muricauda sp. DMG2200308中发现了其他抗氧化剂玉米黄质和β-胡萝卜素的合成,支持了先前关于该属的发现[95,96]。类胡萝卜素通过淬灭高活性单线态氧而成为最有效的抗氧化剂[97,98]。Muricauda sp.菌株GF1产生的玉米黄质通过减少培养的共生菌科(symbiodiiaceae)中的ROS来减轻光和热胁迫[95]。从沿海海砂中分离的Muricauda sp.产生的玉米黄质被证明可以清除一氧化氮[96]。因此,玉米黄质是一种抗氧化剂,可以减轻珊瑚全息体中ROS和RNS的过量产生。
所有选定的候选益生菌都显示出通过norBC还原一氧化氮的潜力,而Roseibium sp. DMG3300306和Pseudophaeobacter sp. DMG2200305也含有这种特性的hmp。此外,除Roseovarius sp. DMG2200361外,所有候选菌株都显示出将一氧化二氮转化为氮的潜力。一氧化氮(norBC)和氧化亚氮还原酶(nosZ)先前在Ruegeria[84]、Muricauda[99]、Roseibium[100]、Pseudovibrio[101]和Roseovarius(仅norBC)[102]中有记载,而在Roseibium sp. DMG3300306和Pseudophaeobacter sp. DMG2200305中发现hmp对这些属来说是新的。通过益生菌减少一氧化氮可能对热胁迫下的珊瑚有利,特别是针对其藻类共生体,因为添加一氧化氮清除化合物可以缓解共生菌科培养物在热胁迫下光合性能的下降[25]。其中3种候选益生菌(Muricauda sp. DMG2200308、P.反硝化菌DMG2200345和Pseudophaeobacter DMG2200305)也显示出通过ahpC将过氧亚硝酸盐还原为硝酸盐的潜力,这是Muricauda和Pseudophaeobacter的新途径。一项早期研究的相关观察结果是,来自与珊瑚相关的水生芽孢杆菌的ahpC可以保护大肠杆菌免受氧化应激[103]。对于假噬杆菌来说,所有四种清除rna的特征都是新的,需要进一步的研究来测试它们的功能。
向珊瑚宿主输出碳,特别是葡萄糖[104]的能力可能是珊瑚益生菌候选物的有利特性,并且可能用于提供能量以增强碳饥饿宿主的漂白耐受性并促进漂白恢复[34,105]。本研究首次报道了珊瑚相关细菌拥有半糖糖蛋白基因,这可能使它们具有像真核同源物SWEET一样输出小糖分子的能力,尽管糖输出在细菌中尚未得到证实[72]。其中,在所选的益生菌候选株Roseovarius sp. DMG2200361、Pseudophaeobacter sp. DMG2200305和P.反硝化菌DMG2200345中发现了SemiSWEET转运体,这是这些属的新发现,表明这些菌株可能向珊瑚宿主输出碳。
为了使细菌益生菌成为增强珊瑚气候适应能力的可行干预手段,必须实现对珊瑚全息剂的长期有益影响[106]。因此,益生菌与全息剂形成稳定的联系是很重要的。在本研究中,我们在4种候选益生菌(Ruegeria sp. DMG2200320、Muricauda sp. DMG2200308、Roseibium sp. DMG3300306和Roseovarius sp. DMG2200361)中鉴定了ARPs和/或WD40-repeat蛋白。据推测,这些elp通过细菌蛋白与真核宿主蛋白的相互作用促进了稳定的共生关系,如一系列与珊瑚相关的细菌(Porites lutea)[107]和珊瑚细菌共生体内生单胞菌(Endozoicomonas sp.)[77]。Muricauda sp. DMG2200308和Roseibium sp. DMG3300306显示出的ARPs和WD40-repeats的数量最多,我们推测这些可能促进了与珊瑚宿主和共生菌科的共生相互作用。在Roseovarius sp. DMG2200361和Pseudophaeobacter sp. DMG2200305中检测到的T4SS也可能通过转运含有锚蛋白重复序列的效应物来帮助它们与珊瑚全息生物结合,这一机制已在一系列细菌中得到报道[40]。一项研究表明,在Roseovarius mucosis中发现的T4SS有助于其鞭毛宿主Alexandrium ostefeldii的定植[108]。T6SS存在于Roseibium sp. DMG3300306和P.反硝化菌DMG2200345中,是细菌中常见的分泌系统,在毒力和抗菌活性方面发挥作用[109],并可能促进细菌的交流[110]。据报道,费氏弧菌中的T6SS通过消除竞争细菌,在与短尾乌贼建立共生关系中发挥作用[111]。珊瑚共生细菌内生单胞菌(Endozoicomonas sp.)中也发现了这种分泌系统[112]。T6SS和T4SS是唯一的分泌系统(包括T3SS),它们可以通过额外的宿主细胞膜运输蛋白质[113],是否T6SS和T4SS在与珊瑚宿主和/或共生菌科建立所讨论的候选益生菌中发挥有益作用还需要进一步的研究。
本研究中分离的一些选定的候选益生菌属已知与珊瑚形成稳定的关联,如Ruegeria spp.[114]。例如,在Pocillopora damicornis的早期和成年阶段都观察到Ruegeria的成员[115]。此外,所选择的一些益生菌候选属与共生菌科或其他藻类密切相关。例如,pomeroyi Ruegeria与硅藻Thalassosira pseudonana形成共生,为其提供必需的维生素B12[116]。共生科和珊瑚不能产生维生素B12[117],但需要它作为中枢代谢酶功能的辅助因子[118]。例如,只要细菌存在,培养的共生菌科(symbiodiiaceae)可以在培养基中不添加维生素B12的情况下生长[119]。因此,所提出的候选益生菌Ruegeria sp. DMG2200320也可能通过为共生菌科和珊瑚提供必需的维生素B12来促进珊瑚全胞菌的功能。Muricauda属、Roseibium属和Roseovarius sp.在培养中与不同的共生体科物种有关联[120]。使用与共生菌科共定位的候选益生菌特别有吸引力,因为ROS和RNS的过量生产主要发生在共生菌科。在最近的一项研究中,从共生菌科中分离出具有高ros清除能力的Roseovarius sp.在接种后的高温下促进了共生菌科的生长(Heric K, Maire J, Deore P, Perez-Gonzalez a, van Oppen MJH:接种Roseovarius增加了珊瑚光共生体Breviolum minutum的耐热性,under review),进一步表明该属菌株可能对珊瑚益生菌有益。
目前的研究增加了珊瑚相关菌株的培养收集和公开可用的基因组。纯培养物对于益生菌接种实验至关重要[121],细菌基因组序列提供了对细菌功能潜力和细菌与珊瑚全息生物相关性的见解。由于束状棘球蚴作为一种新兴的珊瑚模型近年来受到越来越多的关注,该收集将支持旨在建立这一模型的研究。
我们重点研究了通过活性氧和活性氧清除来缓解珊瑚白化的细菌,并提供了从束状菌中分离出来的细菌的假定有益功能的深入列表,其中一些是某些细菌属的新功能。我们对珊瑚相关细菌出口碳的潜力提供了新的见解。每个性状的功能,以及所提出的益生菌菌株对热胁迫下珊瑚全息生物性能的影响,仍需在对照接种实验中进行评估。珊瑚全息剂内候选益生菌的时间稳定性和定位也有待研究。虽然珊瑚益生菌领域仍处于起步阶段,珊瑚全息生物体内细菌的功能尚不清楚,但本研究为确定适合的益生菌菌株提供了重要的一步,旨在建立珊瑚的气候适应能力。
下载原文档:https://link.springer.com/content/pdf/10.1186/s40168-023-01622-x.pdf
为您推荐:
- PDI检测:守护您的家居安全 2025-07-21
- Google Messages在RCS聊天中添加了Ultra HDR图像支持 2025-07-21
- 雪灾图片,雪灾来袭,冰冷冬日中的生命抗争 2025-07-21
- 东契奇,小牛再次输给黄蜂 2025-07-21
- AHA新闻-死因促使法医病理学家发现她有同样的心脏疾病 2025-07-20
- 大学生流媒体交易:查看亚马逊、Hulu、苹果等网站的折扣 2025-07-20


